Computational power in the fight against COVID-19


Computational structural biology is a powerful tool in protein design and engineering. Let’s take a look at how it is being used in the fight against COVID-19. There are several key approaches to solving some of the important problems in structural biology, for example predicting the structure of proteins, as well as predicting how proteins move (e.g., dynamics), and how they interact with other molecules such as drugs (e.g., docking). For protein engineering and design, computational methods have really made a big splash in the last decade, with exciting reports of designing antibodies and other protein biologics as therapeutics. I’ll briefly run through a small selection of these approaches, pointing out examples of how they are being used (or could be used) to design protein therapeutics and vaccines targeting SARS-CoV-2.
Structure prediction based on homology
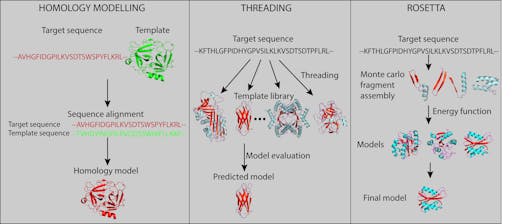
A key contribution comes from a now-mainstream approach of modelling and structure prediction. One of the most widespread techniques is called homology, or comparative modelling. If the sequence of your protein of unknown structure is similar to that of another protein of known structure, its structure can be modelled based on the known structure. A look at COVID-19 UniProtKB shows that there are several viral proteins for which no structure exists (or where only some domains in a multi-domain structure have been solved), and this is where modelling comes in. There are many ways of doing this, for example SWISS-MODEL, which contains a fully annotated database of modelled SARS-CoV-2 proteins. Another powerful and widely-used package, MODELLER, integrates with graphics software such as Chimera and PyMol (via PyMod).
When sequence similarity is too low to allow homology modelling, threading, or energy-based fold-recognition can be used. The I-TASSER server combines threading with an iterative approach of template-based fragment assembly, ab initio modelling, and simulation. The authors have created a resource of the predicted structures of the SARS-CoV-2 proteome, as well as making available 1000s of peptide sequences predicted to block binding between SARS-CoV-2 spike protein and human ACE2.
Over the past two decades software that started life as a structure prediction algorithm has evolved into the full-fledged modelling suite Rosetta. Rosetta spans comparative modelling, folding, docking and design. Rosetta scripts are powerful, but quite a bit of the functionality can be accomplished using ROSIE, an easy to use web front-end.
Prediction from scratch
As the sequence similarity falls below a certain point, sometimes referred to as the ‘twilight zone’ or ~25% sequence identity, it becomes very difficult to accurately produce a model using these methods. However, other methods are available to help here. One of the most widely used methods is Robetta, which has Rosetta at its core, and offers structure prediction by combining homology modelling with ab initio prediction. This approach has been used with great success in general protein design, and marks a gradual and bold departure from laboratory evolution as the most powerful way forward. Rosetta is being used across the board in COVID-19 research, from the design of antivirals, to vaccines. In fact, Robetta was used to accurately predict the structure of the SARS-CoV-2 spike protein several weeks before the structure was determined by CryoEM. Rosetta has an impressive track record when it comes to designing small protein binders as anti-influenza therapeutics, and represents the state-of-the art in combining modelling and experimental testing in massively parallel high-throughput fashion.
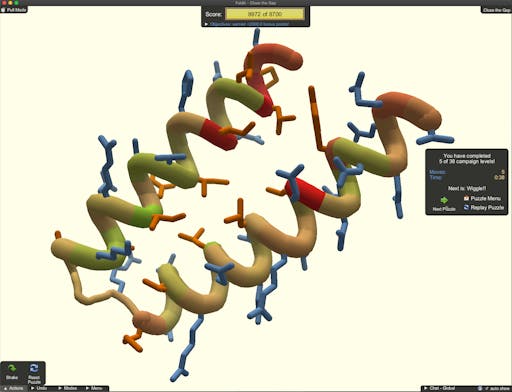
Rosetta has escaped the lab - it is now possible to use this technology by playing the game Foldit. A couple of weeks ago I contributed to an Australian newspaper article on how Foldit is being used to design drugs against COVID-19. One of the successes of Foldit is that it sits on top of Rosetta, but hides the computational complexities from the user - or gamer in this case. So whilst the player is using their creativity at puzzle-solving to get a high score, the underlying calculations ensure that the designed proteins are as physically-realistic as possible. Protein design is a hard problem - designs must be tested in the lab, and the failure rate is high. But, in the case of COVID-19 this is moving fast. Already 99 designs from the first challenge - to design a molecule that could block infection by competing for the spike-ACE2 interaction - are being tested in the lab. The second challenge is underway: design a protein to stop the cytokine release syndrome that may be responsible for severe reactions to SARS-CoV-2 infection, such as respiratory distress.
Artificial intelligence is now making a serious contribution to hard structure prediction problems. DeepMind is using its AlphaFold deep learning system with impressive accuracy, and computational predictions of protein structures associated with COVID-19 is underway. As in the Rosetta approach mentioned earlier, AlphaFold accurately predicted the experimentally-derived structure of the SARS-CoV-2 spike protein.
Comparing methods using blind prediction
In the past 26 years modellers and predictors have come together every year in a competition, to see who’s method is the best at predicting protein structure. The Critical Assessment of protein Structure Prediction (CASP) is an objective way of testing all the methods via blind prediction - experimentalists divulge the sequences of proteins they are determining the structure of, and all best are on - the computational teams then race to predict, and once the experimental work is done, the best prediction, and the winner, is revealed. Prediction of SARS-CoV-2 structures is underway, with some results still coming in from the ~1,600 models submitted by 52 groups.
Predicting protein-protein interactions
Rosetta@Home, another example of citizen science, is being used to great effect in designing binders to SARS-CoV-2 by outsourcing the computationally-intensive step of protein-protein interface redesign to the the vast resource of spare compute power out there - anyone can volunteer their home PC’s to help this effort.
The cousin of CASP, CAPRI (Critical Assessment of PRedicted Interactions) can predict how proteins interact and form complexes. This could be particularly useful in the design of proteins that block infection by SARS-CoV-2. As of writing there are not yet any reports of CAPRI being applied to SARS-CoV-2 proteins.
Proteins are not static, they jiggle and dance. From enzymes to receptors and antibodies, this dynamics is crucial for function. This is apparent on the SARS-CoV-2 spike protein, where structures determined so far represent snapshots - static images of the spike in a particular pose. These snapshots show one of the three receptor binding domains in a receptor-accessible conformation, revealing glimpses into how the spike of the virus engages with the ACE2 receptor on human cells. But to understand the whole event, we need a movie. Doing this in the lab is very tricky and slow, involving combining several methods that can be used to stitch together a lot of data into frames of a movie. But it can be also done computationally using the technique of molecular dynamics simulation (see a discussion of relative merits of experimental and computational techniques for investigating protein dynamics of immune system molecules). Molecular simulation reveals some of the motions that the spike can undergo, but the main challenge here is timescale - using the fastest computers we are limited to simulating for microseconds at best, when in reality the motions are happening over milliseconds and longer - we are 3 orders of magnitude out. But there are two glimmers of home. First, the Anton 2 purpose built supercomputer is the only machine of its type that can achieve such simulation timescale - and DE Shaw, its creators, have just made their simulation data for the spike protein freely available. See some of the movies here. For those interested in the hardware side of things, look here and here.
It’s great to see how different approaches are being combined. A nice example is Michael Feig’s lab who used their prediction pipeline to generate new structure predictions for SARS-CoV-2 proteins that share no known homology with any other structures, subjecting both Rosetta and AlphaFold models to molecular dynamics simulations. This reminds me of the power of making data freely available to all.
As in the case of protein design, distributed computing can be useful here - Folding@home harnesses spare compute power from computers across the globe to perform the molecular dynamics calculations, and is already being harnessed to get a handle on the molecular dynamics of SARS-CoV-2 proteins.
Molecular movies can also be useful designing vaccines and drugs. The choice of a polypeptide antigen in order to make the vaccine depends on the sequence and structural data you have. But if you know that the structure is flexible, you can use the movie to create alternative antigens. Antibody drugs interact with the viral target, latching onto an epitope on its surface. A static snapshot of the viral protein you are interested in may reveal potential epitopes, but what if the protein is changing shape? Potentially important ‘cryptic’ epitopes may only become apparent if you can access the shape-shifting. For example if the spike receptor binding domain needs to alter its shape to open the ACE2 door, knowing how it does that may help the screening or design of drugs that will prevent this.
Enhancing stability
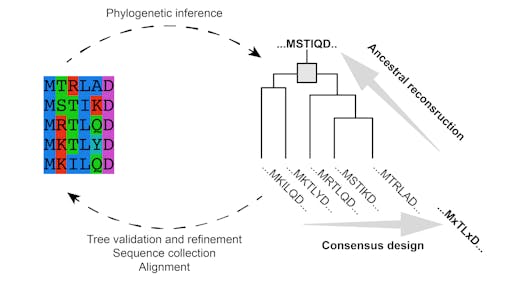
There are many strategies for increasing the stability of a protein. A key problem to solve is to decide which parts of the protein to change. This can be guided by evolution - by looking at the evolutionary sequence variation within the protein family it is possible to identify the conserved residues that likely impart stability, and create a ‘consensus’ sequence, or even re-create a hypothetical evolutionary prototype, as in ancestral reconstruction.
Stability enhancement using Rosetta is expected to play a part in designing ‘mini-protein binders’, targetting the spike protein, and building upon a 2017 study that designed and tested over 20,000 mini proteins targeting influenza and botulinum neurotoxin B, identifying 2,618 high-affinity binders.
In the same vein, these methods can be used to increase the stability and biophysical properties of recombinant forms of the human ACE2 proteins (e.g., for the development of ‘decoy’ molecules that bind the virus spike, thus inhibiting infection), as well as stabilising the viral spike protein, or parts of it such as the receptor binding domain (for use in vaccine development).
A powerful approach that combines the evolutionary, or phylogenetic approach mentioned above with Rosetta-based energy calculations, is PROSS (the Protein One-Stop Repair Shop): its server has been so overwhelmed with jobs involving SARS-CoV-2 related proteins, the PROSS team have pre-calculated the stability-enhancing mutations for human ACE2 and viral spike protein and made them available.
A powerful approach that combines the evolutionary, or phylogenetic approach mentioned above with Rosetta-based energy calculations, is PROSS (the Protein One-Stop Repair Shop): its server has been so overwhelmed with jobs involving SARS-CoV-2 related proteins, the PROSS team have pre-calculated the stability-enhancing mutations for human ACE2 and viral spike protein and made them available.
A powerful approach that combines the evolutionary, or phylogenetic approach mentioned above with Rosetta-based energy calculations, is PROSS (the Protein One-Stop Repair Shop): its server has been so overwhelmed with jobs involving SARS-CoV-2 related proteins, the PROSS team have pre-calculated the stability-enhancing mutations for human ACE2 and viral spike protein and made them available.