COVID-19 Drugs: Inhibiting viral replication and transcription

In a previous post I focussed mainly on large protein molecules as potential therapeutics to treat COVID-19. For example, neutralizing antibodies, antibody cocktails from convalescent patients, nanobodies, and non-antibody scaffolds such as FN3 monobodies that could be used to block infection. To fight COVID-19 in the short term, for example treating patients who have severe symptoms (perhaps lowering the burden on intensive care units), we need drugs that can treat infected people. In this post I want to discuss small molecule drugs candidates undergoing clinical trials, and how structural biology can play a crucial part in the spectrum of strategies for antiviral drug design.
There are already many drug candidates in the pipeline, including existing drugs that can be repurposed. In late March 2020 the WHO launched a global trial, called SOLIDARITY, to investigate whether various existing drugs can treat COVID-19. SOLIDARITY is testing three treatments: remdesivir (a broad-spectrum inhibitor of RNA viral replication) and two combinations of ritonavir, lopinavir (both HIV protease inhibitors) and interferon β1a (an endogenous immune activator). This is in addition to over 1900 COVID-19 - related clinical studies currently in progress looking at a very broad range of treatments. The RECOVERY trial in the UK, enrolling over 11,000 patients from 175 NHS hospitals, is also testing a range of drugs including the steroid anti-inflammatory dexamethasone and antibiotic azithromycin. These trials are on-going, except for one: the trial of hydroxychloroquine has been terminated because it found no beneficial effect in patients hospitalised with COVID-19.
The lifecycle of SARS-CoV-2 reveals several key processes performed by enzymes that could be targeted by repurposing existing drugs. Lets look at how drugs can inhibit a key early stage in the viral lifecycle - replication and transcription. Similarities between mechanisms of viral replication mean that drugs developed to target SARS-CoV-1 and MERS-CoV could also be effective against SARS-CoV-2.
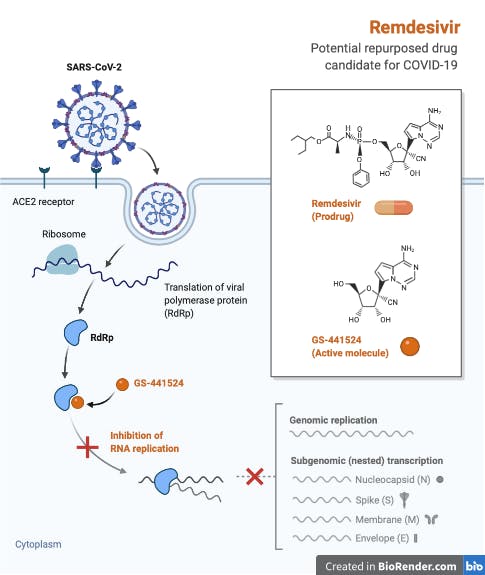
Originally trialled, unsuccessfully, in a 2019 outbreak of Ebola in the Democratic Republic of Congo, remdesivir is a nucleotide analog that mimics adenosine, thus targeting viral replication by binding, and inhibiting RNA-dependent RNA polymerase (RdRp). A detailed discussion of the discovery and development of remdesivir can be found in this excellent review. Preliminary results of a double-blind, randomized, placebo-controlled trial of intravenous remdesivir in adults hospitalized with COVID-19 with evidence of lower respiratory tract involvement have just been published, showing that remdesivir was superior to placebo in shortening the time to recovery. This is significant as it is the first drug to be recommended for treatment - the Food and Drug Administration made remdesivir available on May 1st 2020 under an emergency-use authorization for the treatment of severe COVID-19.
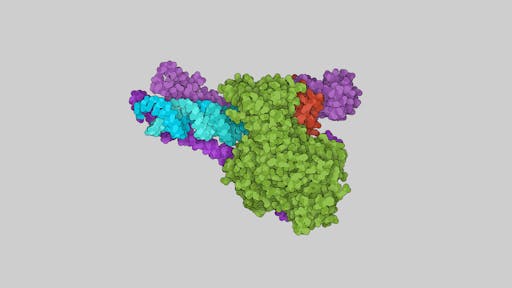
SARS-CoV-2 replication machinery
Structure determination of the remdesivir's target enzyme, the RdRp of SARS-CoV-2 is a prime example of structural biology’s key role and how multiple international teams all made rapid progress: it was one of the first viral enzymes to be solved, by Rao’s group in Beijing/Shanghai using CryoEM, revealing the structural architecture of this key enzyme in the virus transcription machinery, including the conserved active site.
The structure from Rao's group allowed remdesivir to be modelled in the enzyme active site, giving clues to its inhibitory potential. Things continued to move very quickly - Patrick Cramer’s team in Göttingen have just reported the structure of SARS-CoV-2 RdRp in its replicating form. In another example of the power of making data freely available, Cramer's team made use of the Rao's structure of free SARS-CoV-2 RdRp (PDB: 6M71) that was 'tweaked' by Tristan Croll. The team were also able to use Rao's 2005 crystal structure of the nsp7-nsp8 hexadecamer from SARS-CoV to model the structurally similar helical segments in the N-terminal regions of both copies of nsp8. I have used the RNA-bound structure from the Cramer lab to illustrate some key features of the enzyme below. One of the neat features of this new structure are long helical extensions in nsp8 that were disordered in previous structures. The helical extensions, running parallel to the exiting RNA duplex, make charged interactions with it, forming positively charged 'sliding poles' that may help to guide the RNA as it exits. The Cramer lab page has some nice movies showing how the RdRp enzyme works.
Remdesivir disrupts viral replication
Xu’s team in Shanghai also report the structure of the apo enzyme (e.g., no RNA bound) as well as that bound to a 50-base template-primer RNA and remdesivir. Like many nucleotide analog prodrugs, remdesivir is incorporated into the nascent viral RNA by the error-prone viral RdRp, thus disrupting RNA synthesis through delayed chain termination, or via the accumulation of deleterious mutations in the viral genome. That requires the conversion of the parent drug to the active triphosphate form: remdesivir is a prodrug that is intracellularly metabolised to the active, triphosphate drug form (RTP, closely resembling ATP). In the Xu paper, inhibition of RNA polymerisation activity of RdRp was achieved only using the RTP form of remdesivir, and not the prodrug or the monophosphate (RMP) form. The complex structure from Xu's team contains 14-bases of RNA in the template strand, 11-bases in the primer strand, and the inhibitor remdesivir in the monophosphate form covalently linked to the 3' end of the RNA primer strand. The active site also contains a pyrophosphate molecule and two magnesium ions that may play a role in the catalytic mechanism.
Consistent with the evidence for a chain termination mechanism of action of remdesivir, only a single RMP molecule is assembled onto the RNA primer strand, despite an excess of RTP in the sample used for structure elucidation. Whether remdesivir functions as an 'immediate' or 'delayed' RNA chain terminator must await mechanistic studies, though the Cramer team suggests that 'delayed' termination explains how remdesivir can escape removal from the RNA 3'-end by the viral exonuclease nsp14. That is, incorporation of remdesivir into the nascent RNA chain does not result in immediate termination, which is delayed until several more nucleotides are added to the RNA. Below is a simple animation from the Cramer lab showing NTP's being added to the growing RNA chain during replication.
Remdesivir makes a number of interactions at the active site, all mediated by its base and consistent with its role as a nucleoside analog (see slideshow above). Specifically, these are; base-stacking with the upstream base from the primer strand, two base-pairing hydrogen bonds with the uridine base from the template strand, and polar contacts with the sidechains of K545 and R555, which the authors suggest may act to stabilise the incoming nucleotide in the correct position for catalysis. The phosphate diester backbone of RMP coordinates two magnesium ions and the pyrophosphate, which the authors suggest may block the entry of nucleotide triphosphate to the active site channel (this can be seen clearly in the last two slides).
There are now three reported structures of the SARS-CoV-2 RdRp: free/apo, RNA-bound, and in complex with RNA+remdesivir. All three of these structures, determined at breakneck speed, were solved using CryoEM. What's more, the resolution and quality of the data is very impressive, revealing key insights into how remdesivir - the only licensed drug to combat COVID-19 - inhibits the enzyme, as well as new information showing how previously disordered helical regions of the enzyme stabilise the exiting RNA duplex. This is further evidence of the impressive progress of the "resolution revolution" in CryoEM, and the technique's game-changing effect on the structural biology landscape. In a previous post I discussed the importance of making raw data available - it is great to see the RdRp-RNA-remdesivir complex data from the Cramer lab deposited here.
Mechanistic insights and drug design
The availability of three structures has further advantages. Despite some interesting structural differences between the apo and RNA-bound structures (for example a rearrangement of the nsp7-nsp8 interface, and movement within the thumb subdomain to accomodate the duplex RNA), the structure of RdRp active site is relatively conserved, which may allow the design of broad-spectrum antiviral inhibitors. To explore this further, Xu's team used their model to dock in the structures of other nucleotide analog drugs, including favipiravir, ribavirin, galidesivir and EIDD-2801, which all inhibit SARS-CoV-2 replication in cellular assays. The proposed mechanism of these nucleotide analogs is the same as remdesivir, e.g., inhibiting the viral RdRp through RNA chain termination. Docking of EIDD-2801 into the active site reveals two extra hydrogen bonds compared with remdesivir, one with the RNA and one with the protein. This is consistent with the higher potency of EIDD-2801 in inhibiting SARS-CoV-2 replication. These models haven't been validated or made available, but details can be found in the SI section of their paper.
Two collaborating groups at Colorado state and CNRS Marseille have recently reported an interesting study (preprint, currently undergoing peer-review) investigating the use of the influenza drug favipiravir (a guanosine analog), as a promising inhibitor of SARS-CoV-2 RdRp. The authors show that once incorporated into viral RNA, favipiravir acts as a mutagen that can escape the viral repair machinery, and suggest that the drug may exert additional pressure due to the relatively low cytosine content of the CoV-2 genome. In addition, they reveal data that show the SARS-CoV RdRp to be the fastest viral RdRp known, which may be necassary to rapidly replicate its ~30,000 nucleotide long RNA genome, but at a cost to replication fidelity. Using the CryoEM structures of the SARS-CoV-2 RdRp, the authors propose an intruiging explanation for the molecular basis for these observations. As mentioned above, the active site of most RNA virus RdRp enzymes anchor the catalytic R555 in a conformation that will catalyse the correct incorporation of NTPs, using a salt bridge between it and E547. In the RdRp of SARS-CoV-2, however, this salt bridge is absent because residue 547 is an alanine; this may allow flexibility of R555 and thus relaxed triphosphate positioning and base-pairing, ultimately lowering the RdRp fidelity (see slide 8 in the animation above).
These structures provide a solid rationale for understanding how these drugs disrupt the SARS-CoV-2 RdRp activity and thus inhibit viral replication, and a starting point for modifying the existing nucleotide drugs, including the highly potent EIDD-2801. Structure-based drug design is a long road with many obstacles, but can lead to some game-changing outcomes. One of the best examples is that of HIV protease inhibitors. After structure determination of HIV protease in 1989, licensed drugs appeared in the clinic six years later, with 10 drugs currently in clinical use as part of multi-drug antiretroviral therapies.
It is worth remembering that remdesivir was originally developed as an antiviral drug to combat Ebola, but was found to be less effective than antibody-based therapeutics, which had a greater effect on lowering death rates. This underlines the need to rigorously pursue the development and testing of many drugs having different mechanisms of action, as it is highly likely that future effective treatments will involve a combination of antivirals and other therapeutics. This is exemplified by current HIV treatments, which consist of patient-specific combinations of drugs that inhibit HIV protease, integrase, and reverse transcriptase enzymes, among others.