Engineering T cell receptors for effective T cell activation and immunotherapy

In my field of study, I investigate how T cells become activated through recognition of foreign peptides from viruses or cancers. Our cells naturally digest proteins and present these protein fragments or peptides (p) on top of Major Histocompatibility Complex (MHC) molecules on the cell’s surface. T cells use their unique T cell receptors (TCRs) to recognize and bind to these peptide complexes (pMHCs), where the quality of binding influences T cell activation, but its exact mechanisms are still a little hazy. Activated T cells kill infected cells and allow the immune system to successfully control and fight infection.
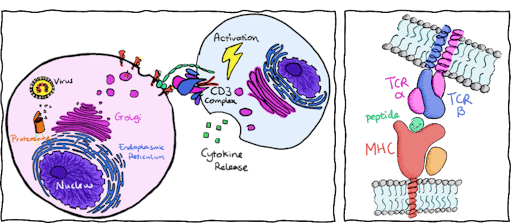
To harness this natural defence mechanism, adoptive T cell therapies aim to engineer T cells to specifically target and activate against viral or cancerous peptides. Although the atomic structure of hundreds of TCRs and pMHCs are available, we still have not worked out an effective way to trigger T cell activation. T cells usually activate and bind to pMHCs with a binding affinity within the micromolar range (10-4 to 10-6 M). Yet engineered T cells, that focus on enhancing binding affinity to the nanomolar range and above (10-7 to 10-9 M) can still fail to activate. What’s more, engineering TCRs with high affinity to cancer targets can have dangerous consequences. Cancerous peptides look very similar to host-cell peptides and increasing affinity towards cancer peptides could lead to a high risk of off-target activation.
It is still not clear what the determinants of activation are, but we know why increasing affinity between TCRs and pMHCs does not always work. This is because:
- T cell activation is the result of a cell-to-cell interaction and activation occurs after a threshold of net binding. The expression of T cell receptors and pMHCs on the cell surface is affected by many factors including protein stability, peptide abundance, and peptide affinity.
- The way binding affinity is measured and reported for T cell activation is too simplistic. For many T cell activation studies that measure TCR-pMHC binding, most only report the dissociation constant and not the kinetics of binding, leaving out crucial information such as association and dissociation rates and bond lifetime.
Simply put, T cell activation is different to the 1:1 binding affinity models typically used for drug-receptor interactions. Despite this, the reigning hypothesis for the past 30 years is that high affinity between the TCR and pMHC leads to T cell activation. More recent studies, especially bioforce probe experiments, show strong support that binding avidity better corresponds to T cell activation as it measures binding at the cellular level (taking into account expression levels, protein stability, co-receptor binding, and more). Instead of focusing purely on improving affinity, we need to investigate the precise mechanisms that enable activation. A good example is this recent publication of how binding avidity is being used to inform the design of T cell receptor engineering for adoptive T cell therapy.
What we know about T cell activation
Currently, there are two models of T cell activation: the kinetic proofreading model and the serial triggering model. The kinetic proofreading model proposes that binding at the cell-cell interface must have a net bond lifetime of at least 2 seconds, long enough for downstream signaling of T cell activation to occur. Against this, the serial triggering model argues that as long as enough TCRs continuously bind to pMHCs, regardless of duration, then activation can be achieved. In favour of this model speaks that we know that TCRs that bind with a fast association rate, fast dissociation rate, and short bond lifetimes can indeed activate T cells. In short, kinetic proofreading is like a press and hold type of switch; whilst serial triggering activates when you press it as fast as you can.
Personally, I am not so fond of serial triggering and here is why: Although there are T cells that activate through serial triggering when tested in the lab, these studies use concentrations of pMHCs that are beyond what we might find in a cellular context. Additionally, serial triggering does not take into consideration co-receptor binding synergy, in which the CD8 co-receptor has been proven to play an enormous role in T cell activation.
Disulfide bond model in TCR-pMHC
In my most recent work, I engineered a disulfide bond between a TCR and its target peptide. Initially, this study began with an observation that the TCR region (CDR3 loops) that primarily interacts with the target peptide rarely possesses cysteine residues. This is likely due to central tolerance, a process in the thymus which screens and deletes immature T cells that react too aggressively to self-peptides. A free cysteine in the CDR3 region may potentially form disulfide bonds and cause unwarranted activation.
To make a working model for this study, we introduced cysteine residues into the CDR3 loop of T cells and found that they differentiate into intestinal epithelial T cells that reside in the gut lining. To mimic and study this sort of disulfide bond, I introduced cysteine’s between the TCR and target peptide and was able to use X-ray crystallography to verify its structure at a high resolution of 1.9 Å.

I used surface plasmon resonance (SPR) to characterize the binding of this system and found that when bound, the TCR and pMHC do not dissociate away from each other. Inadvertently, we have created a covalent disulfide-linked TCR-pMHC system, where the TCR-pMHC bond lifetime is prolonged indefinitely and could be a fantastic way to uncouple the parameter of bond lifetime from the two models of T cell activation.
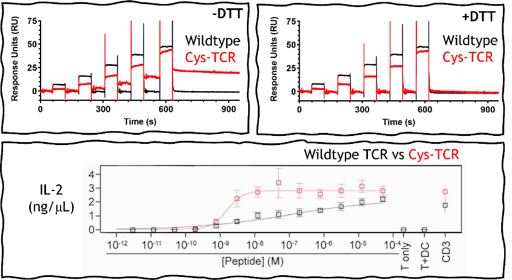
When we put this disulfide bond TCR-pMHC into real-world biology by transducing these TCRs into mice T cells and introduced the cysteine-peptide, these T cells were shown to be 50-fold more responsive to activation than the wildtype system. T cell activation was detected at picomolar concentrations of the cysteine-peptide. Importantly, our binding experiments also revealed that the formation of the disulfide bond did not change binding affinity of the TCR when compared to the wild-type system. This suggests that unlike high-affinity TCRs, they are less likely to lose specificity or induce off-target activation and toxicity. I also tested binding against other pMHCs that contained exposed free cysteine residues, and found no off-target disulfide bond formation indicating that TCRs must first be able to recognize and bind to the pMHC before the disulfide bond can form.
Together, this approach is ideal as a novel design for T cell engineering in adoptive T cell therapy. This is because of:
- Increased T cell sensitivity and activation
- No change in binding affinity for the TCR
- No off-target binding through non-specific disulfide interactions
Of course, a limitation for this approach would be that the neoantigen or cancerous peptide would need to possess a cysteine mutation. This work has just been published in Nature Communications and this type of TCR modification is patented with Monash University, Australia. The team and I are still at the preliminary stages of advancing this technology. Sometimes it amazes me how we could dive head-first into engineering T cells for therapeutics without firstly understanding the parameters that successfully activate T cells. Therefore, we also plan to employ this unique system to interrogate how extended bond lifetime affects T cell activation in both serial triggering and kinetic proofreading models of activation, and hopefully uncover the secrets behind T cell activation.