Machine Learning for Protein Engineering - here to stay

Machine Learning and Deep Learning have been buzzwords for quite some time now, but in the last few years, the impact on real-world applications has started to show. There has been much excitement around the AI-powered predictions of protein structures made by the AlphaFold Protein Structure Database. But what kind of machine learning is behind AlphaFold and other applications? Why has recent progress been so rapid and what will the future hold?
ML101: What is Machine Learning?
Machine Learning (ML) is only one approach in the broader field of Artificial Intelligence. There are many definitions for ML, but I especially like this one:
“A branch of artificial intelligence (AI) and computer science which focuses on the use of data and algorithms to imitate the way that humans learn, gradually improving its accuracy.” - IBM
A machine learning approach is increasingly useful in the life sciences due to both the sheer complexity of living systems and the exploding amount of data produced by methods such as next-generation sequencing and genetic testing.
Deep Learning is a subfield of Machine Learning which was born (some might say reborn) in the 2000s when advances in hardware and data availability methods that had been previously theoretical meant Deep learning could now be applied in practice.
For ML we distinguish between three main categories of learning: supervised, unsupervised, and reinforcement learning. Supervised and unsupervised learning, and their intermediate, self-supervised learning, have had wide-reaching impacts on the protein engineering community.
Supervised Learning: Watch and Learn
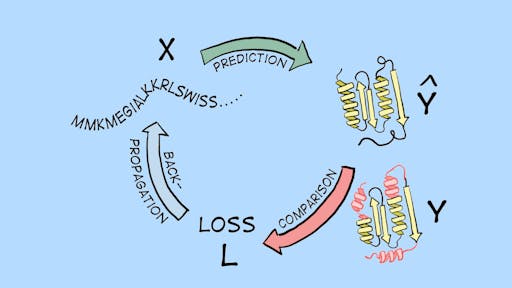
Supervised Learning describes the process many of us imagine when we first hear about Machine Learning: I have some input data X and want to predict an output Y. This was the subfield in which Deep Learning showed its powers, first in datasets such as MNIST for handwritten digits or ImageNet for images. These datasets are used to compare and test newly developed ML algorithms and are not current “real-world” applications driven by ML.
In these settings, the algorithm is given a labeled data set with training examples (for which it knows both input X and output Y) and is trained to produce new outputs Y for as-yet-unseen input X.
The most famous example of applying this type of learning to protein engineering is AlphaFold2: while it is a complicated model with different kinds of inputs/outputs, in principle it was trained on protein sequences (input X) to predict 3D protein structures (output Y) (see Fig. 1).
This approach had quite some success in the past, but it has one fundamental limitation: the algorithm needs labeled data in the first place in order to learn patterns and make predictions later. Often, we do not have the luxury of labeled data or it is limited/expensive; When engineering proteins, unlabelled data in the form of protein sequences is abundant but labeled data in the form of protein structures via experimental procedures such as X-Ray crystallography/cryoEM is very expensive and thus less available. The development of AlphaFold2 is changing this, as it now provides us with reasonable structures for most known proteins, but there are still specific cases such as orphan proteins without known homologs or newly designed sequences where other models challenge AlphaFold2 by taking inspiration from another learning paradigm: unsupervised learning.
Unsupervised Learning: Figure it out yourself
Unsupervised Learning in contrast does not rely on labeled data and mimics how we as humans learn: it relies on recognizing patterns internal to the unlabelled data to solve tasks, for example by arranging data points in different categories (clustering) or representing data in a simpler form (dimensionality reduction) (Fig 2).

Often, self-supervised learning techniques are subsumed in the unsupervised learning category. It also deals with unlabelled data, but we trick the system by using part of our input as the output labels that we are trying to predict. In our case of protein structure prediction, an unlabelled protein structure is one for which we do know the input (sequence) but not the output label (structure). In this case, we cannot simply train a supervised learning algorithm as explained above with it since we do not know the true output (structure) to begin with!
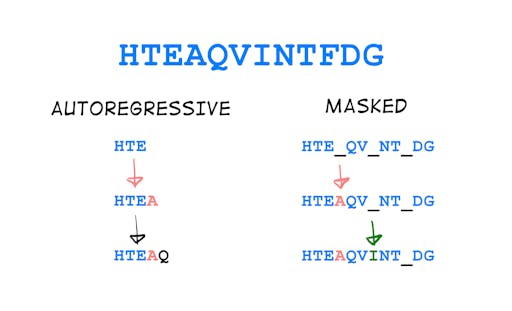
But self-supervised learning gives us tools to still learn something about the sequence. For example, in the case of an unlabelled protein sequence, we could predict the residues one after the other (autoregressive modeling) or hide some residues from the model and ask it to fill in the blanks (masked language modeling; see Fig. 3). With this procedure, the model does not know about the output label (structure), but gets familiar with the properties of the input (sequence) and gets a “feel” for it, which enables it for example to generate new sequences that have desirable biophysical properties such as being easy to express or thermostable.
This approach has been immensely successful in protein engineering in the last few years. Recently, a special form of this method using language models has taken the world of biology by storm. Originally developed for use cases such as language translation with DeepL, protein-specific language models such as ProtGPT2 or ESM-1b make use of the millions of unlabelled sequences at our disposal. These models can be used to predict effects that changes in the sequence might have such as increased binding affinity (EMS-1v), or even try to design proteins by predicting sequences from structure (ESM-IF1).
One interesting phenomenon of large language models is that when scaling them larger and larger, they exhibit not only better performance as expected, but also show some surprising new features called emergent abilities. This can be for example the performance of arithmetic calculations for NLP models such as GPT3 or the prediction of 3D structure in the case of protein language models. This recently enabled MetaAI to publish the ESM Metagenomic Atlas, a resource with more than 600 million predicted protein structures (for comparison, the AlphaFold database contains around 200 million predictions). Due to the speed of the prediction, you can even fold your sequences now online! This comes with a speed-vs-accuracy trade-off but is still a very exciting development in the field. With this powerful resource in their hand, structural biologists and protein engineers can quickly test out hypotheses about their proteins of interest and iterate at a much faster pace to deliver new designs and insights, pushing the frontier of protein science and drug discovery even further.
Back to reality: What has AI achieved so far in biology?
The triumph of AlphaFold2 in predicting protein structures definitely had the broadest media coverage. And it will certainly have a huge impact on the way research in this area is going forward. So far, most of the achievements that AI has made in the field of protein engineering are still in the stage of research publications and are not yet available to patients as clinical candidates.
That being said, companies such as Exscientia are already showing that the superiority of AI algorithms over humans does not only play out in chess and Go but can also help in drug design. Numerous start-ups are working on AI approaches to biology with record funding available. And with the immense AI-powered progress in other fields, it would not be surprising if protein engineering in a few years will look very different from how it does today!
Challenges: Where does AI struggle?
Despite recent successes and bright outlooks, there are still several topics in protein engineering that AI struggles with. For example, predicting binding affinities from protein-ligand structures (of interest for virtual screening applications in drug discovery) does not seem to benefit much from current deep learning methods.
Another category of targets that gained increasing attention in the last decade is intrinsically disordered proteins. Here, AlphaFold2 seems to be well-suited at predicting disorder, while it fails to predict protein fold switching, a phenomenon in which a protein occurs in two or more stable conformations that are interconvertible. Disordered proteins occur often in nature and proteins often switch states when performing biologically important functions such as binding to receptors, so future advances in these areas will be important to leverage the full potential of deep learning for protein engineering.
Finally, while huge progress has been made in predicting structures for almost all known proteins, the folding process describing how they end up there is still not fully understood. There is evidence that models such as AlphaFold2 may help us in understanding this process, but currently, their trajectories do not have much in common with other parameters known to describe protein folding processes. This may be due to the difference between the process of in-silico and in-vivo co-translational folding or even to current shortcomings in the understanding of the underlying energetics. Either way, there is lots of work left to be done!
Conclusion: What might the future hold?
Currently, we are seeing new preprints about ML for protein engineering daily, with both industrial and academic research focused on propelling the field forward. While current design pipelines in industry rely heavily on experimental procedures and physics-based modeling, their performance can be improved by combining these approaches with deep learning and even replacing them with novel ML-based methods in some instances. These advances could decrease the cost and time needed for protein engineering by several orders of magnitude and reduce designing proteins for common use cases to a simple click of a button in a few years’ time.
As mentioned above, there are still a number of things we do not understand and cannot do with the current methods, but with the number of resources and brain power currently invested in the topic, it will not be long until this post will need an update!
Learn more about Kieran at his website.