SARS-CoV-2: getting the data out there, and getting more from it


In addition to providing a gold mine of insights into the function of many viral proteins (which will allow structure-based drug design – more on that in another post) in a staggeringly short timeframe, the processed X-ray diffraction data used to produce the structural models are freely available (e.g., at the PDB). This means that anyone is able to improve these structures using open source software. Better structures mean more biological insights. I’m not suggesting of course that there are problems with any of the deposited structures, rather that, as structural biologists we accept that model building and refinement relies on the tools currently available – as better tools emerge, it is always possible to repeat the interpretation of the electron density map and also the model refinement. This increases the biological insights that can be potentially gleaned from the model. It’s great to see that in the last few days, reports of re-refinement of SARS-CoV-2-related structures have emerged. What’s more, the methods developers have come together to create the Coronavirus Structural Taskforce, a repository of data, structures and analysis that will allow the methods to squeeze every bit of biological insight out of the datasets.
Raw Power

If the raw experimental data (unprocessed diffraction images) are available, there is the potential to push the quality and completeness of these structures even further - even possibly extending the data resolution in cases for example where improved software can process reflections too weak to be measured in the original processing. Articles discussing the why’s and the how’s of raw X-ray crystallographic data availability can be found here:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4187998/
- https://journals.iucr.org/d/issues/2016/11/00/yt5099/
- http://journals.iucr.org/m/issues/2017/01/00/ti5008/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6874509/
- http://journals.iucr.org/d/issues/2008/07/00/gx5131/
In some cases, diffraction data either cannot be processed or are too problematic to allow structure refinement to proceed, and hence structures do not emerge – this is a strong argument that making raw data available in these cases could prove the difference between a structure or no structure (e.g., biological insights or not). The vast majority of X-ray crystallographic data originates at synchrotrons, and it is great to see that many around the world are already providing COVID-19 resources. E.g., Diamond UK, and DOE light sources in US. Over the past 10 years sharing raw diffraction data has gathered momentum, with several cloud based services now available, for example the Integrated Resource for Reproducibility in Macromolecular Crystallography, Zenodo, and the SBGrid Data Bank.
Things are happening fast - protein crystallographers at the Diamond synchrotron (UK) yesterday uploaded 78 raw diffraction datasets for structures of the SARS-CoV-2 main protease in complex with a range of small molecule inhibitors/ligands.
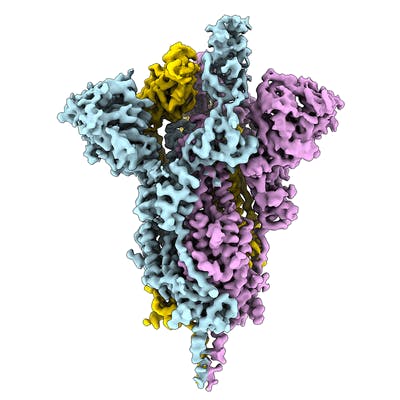
Electron microscopy (EM) is playing an important role in structure determination these days, for example the structure of the SARS-CoV-2 spike ectodomain structure (open state) was determined by EM. The EMDB page for that entry allows anyone to download the map that was used to fit and refine the atomic coordinates. Over the coming days and weeks this resource is likely to grow.